| The
POOL Lab Research People Publications Genomes |
| Analysis of Population Genomic Data Population genetics has recently undergone a profound transformation as sequence data sets expanded from the single gene scale to the whole-genome scale. Once a primarily theoretical field limited by scarce empirical data, population genetics is now experiencing an avalanche of data as large numbers of sequenced genomes start to be published. Just as the initial publication of full genome sequences revolutionized the field of molecular genetics, genomic data now gives population genetics the unprecedented resource of a “complete data set” on genetic variation. Such data may help us address some of the most important and long-standing questions in population genetics, such as: What is the relative importance of natural selection and population history in determining levels of genetic diversity across the genome? And, what types of mutations are the raw material of adaptive evolution? Our ability to fully harness the potential of population genomic data is currently restricted by a lack of appropriate statistical and computational methods that are applicable to genome-scale data and take advantage of its unique features. Part of our lab's research centers on the developing novel analysis methods for population genomic data and applying these methods to obtain biological insights from such data. 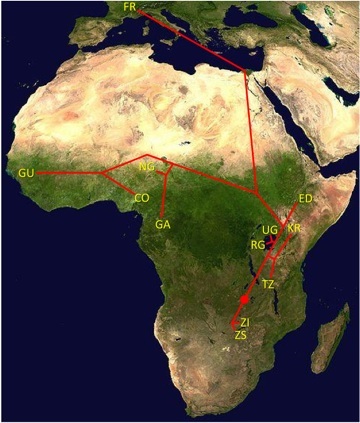 A good example of the applied side of our interest in population genomic data comes from a recent analysis of >100 sequenced genomes from sub-Saharan populations of Drosophila melanogaster (Pool et al. 2012 PLoS Genetics). Here, we used genomic variation to learn about the history and genetic structure of this species within its African ancestral range, and identified genes likely targeted by natural selection based on genetic variation within and between populations. We also documented two cases where natural selection drives rapid intercontinental gene flow, with consequences for genome-wide diversity: widespread gene flow back into Africa (perhaps associated with adaptation to urban environments) and the recent African origin of inversion polymorphisms that are now common in Europe. Moving forward, one of our interests is in developing new methods that allow natural selection and population history to be studied simultaneously. It's well appreciated that non-equilibrium demographic history can interfere with inferences of selection, and so conventionally, population history is estimated first. However, natural selection will be especially effective in the genomes of organisms with large population sizes. Published evidence suggests that for D. melanogaster, the typical assumption that random sites are rarely affected by hitchhiking may be unwarranted, and demographic estimates obtained under neutral assumptions may be inaccurate. Hence, we may need to make joint inferences about demography and selection in order to understand either process. Other interests in the development of population genomic analysis methods include: * Improving the use of haplotype information for identifying adaptive population differences. * Integrating population genetic signals of selection with genotype-phenotype association data. * Developing a population genetic framework for gene regulatory data from RNAseq.  |